No espaço de poucas semanas, o interesse mundial sobre virologia, epidemiologia, entre outros temas biológicos, ultrapassou os ambientes acadêmicos e hospitalares para se estabelecer entre a população como um todo. Todos ouvimos ou lemos na grande mídia de massa, tradicional ou online, sobre o COVID-19 e o vírus que o causa: SARS-CoV-2. Notamos um aumento da relevância desses termos nas conversas entre amigos e nas redes sociais, ocasionando também um aumento de rumores, desinformação e de fake news.
Desde o início do surto na cidade de Wuhan, da província de Hubei na China, e o estabelecimento de uma pandemia, o número de artigos científicos sobre esse vírus vem aumentando. Um total de 978 resultados podem ser encontrados hoje, 05.04.2020, em uma pesquisa das palavras-chaves (COVID-19) AND (SARS-CoV-2) no PubMed. Mas esse número se torna muito superior, graças aos artigos publicados em revistas não-indexadas pelo PubMed, incluindo artigos regionais (como os publicados apenas em língua chinesa) e pre-prints.
No entanto, mesmo com o aumento de informações em diferentes áreas do conhecimento relacionadas com o combate desse patógeno, ainda existem muitas áreas cinzentas, principalmente com relação às suas origens.
Inúmeras perguntas científicas surgiram ao longo desse período. Em quais espécies animais o vírus surgiu? Em morcegos, em um pangolim ou outra espécie selvagem? Será que ele apresentou apenas um hospedeiro ou contou com um hospedeiro intermediário? Onde surgiu? De uma caverna ou floresta na província chinesa de Hubei ou de outra localidade? A transmissão tem relação com os hábitos alimentares dos chineses?
Além das perguntas científicas válidas, algumas hipóteses pautadas puramente em teorias da conspiração também surgiram e foram bastante difundidas em redes sociais, incluindo a de pessoas famosas e políticos ao redor do mundo, o que inclui o Brasil. Será que esse vírus foi criado pela China com interesses econômicos? Será que foi produzido em laboratório e não tem uma origem natural? Será que teve origem natural, mas escapou ou foi solto propositalmente de um laboratório? Hipóteses fantasiosas e que serviriam como roteiro para um novo filme de ficção científica explodiram no Whatsapp e se tornaram lendas urbanas em pouco tempo. E nem vamos contar as explicações de caráter religioso, quase sempre punitivas.
Pensando nisso, esse texto tem como objetivo compilar algumas informações sobre a origem do SARS-CoV-2, responsável pelo COVID-19, além de apresentar e discutir alguns métodos de bioinformática empregados para estabelecer essa origem. Se o seu interesse é apenas saber a origem do SARS-CoV-2, já adianto, a resposta não é tão simples quanto alguns estão tentando fazer parecer.

Surgimento do COVID-19
Tudo começou realmente em 2019. Especificamente, em dezembro de 2019, 27 das primeiras 41 pessoas hospitalizadas com uma nova doença respiratória de origem viral (66%) passaram por um mercado localizado no coração da cidade de Wuhan, na província de Hubei. Essa característica e coincidência poderia ser nossa primeira evidência para tentar buscar uma correlação entre o vírus adquirido e o mercado, mas como sempre temos que lembrar: correlação não implica causalidade.
Com isso, de acordo com um estudo realizado no Hospital Wuhan, sabemos que o primeiro caso humano identificado não havia frequentado esse mercado e teria surgido em novembro de 2019. Para fortalecer ainda mais essa ausência de causalidade e a data de início dessa epidemia, e corroborar as informações noticiadas pela mídia chinesa e pelo governo, uma estimativa de datação molecular baseada nas sequências genômicas do SARS-CoV-2 indicou que essa cepa teria se originado realmente em novembro daquele ano. Além disso, com base na baixa variabilidade verificada entre as sequências genômicas conhecidas do SARS-CoV-2, se estabeleceu um consenso de que a cepa viral tenha sido detectada pelas autoridades poucas semanas depois de ter emergido entre a população humana no final de 2019 [*,*]. Essas características foram importantes para que os pesquisadores levantassem questões sobre o vínculo entre a epidemia emergente do COVID-19 e a vida selvagem local [*].
Genoma Sequenciado
Com o descobrimento da doença e a rápida expansão dessa epidemia emergente, o primeiro genoma do SARS-CoV-2 foi rapidamente sequenciado por pesquisadores chineses.
Sobre o processo de montagem desse genoma, em um momento de epidemia emergente, preciso fazer algumas considerações. Em primeiro lugar, devo realmente parabenizar os autores (Ok, eles não vão ler esse texto, mas vale a intenção). Todos os dados brutos de sequenciamento foram disponibilizados na base de dados SRA do NCBI e seus identificadores fornecidos, promovendo assim o princípio de Open Data e Open Science.
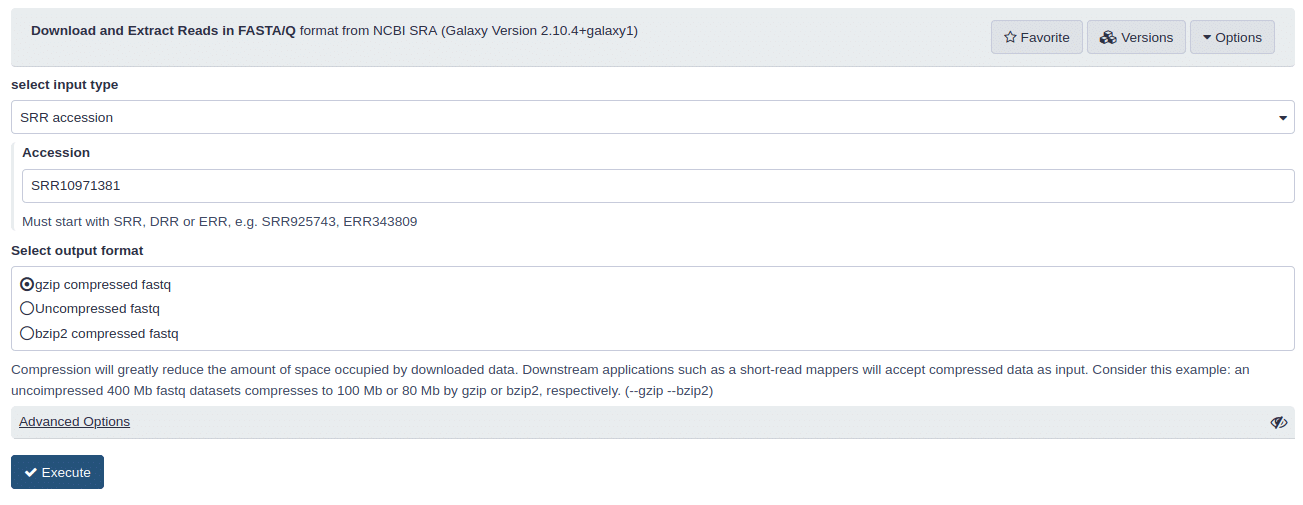
Com as bibliotecas que saem do sequenciador (nesse caso um Illumina MiniSeq) disponíveis de forma pública, qualquer pessoa interessada (pesquisador ou não) poderia reutilizar as bibliotecas para novas análises. Pensando nisso, utilizei o ID SRR10971381, referente a esse sequenciamento, para realizar o download desses dados brutos, através da ferramente Download and Extract Reads in FASTA/Q format from NCBI SRA, para o meu usuário da aplicação de bioinformática Galaxy, em uma instância européia (https://usegalaxy.eu/).
Além de fornecer os dados brutos de sequenciamento, os autores também descreveram de forma detalhada todas as etapas realizadas durante o processo de montagem do genoma viral, além de utilizarem apenas ferramentas de domínio público e open source, o que permite a reprodutibilidade e replicabilidade de toda essa etapa durante a revisão do artigo ou por qualquer outro interessado. Segundo os autores, os mesmos utilizaram os softwares MEGAHIT e Trinity (ambos disponíveis no Galaxy) para a montagem dos contigs, depois reduziram a redundância das sequências através dos softwares BLAST e Diamond, removeram as sequências do hospedeiro (humano) através do Bowtie2 e, por último, realizaram uma análise de abundância de contigs virais baseado no RSEM. Em resumo, todos softwares gratuitos e de livre acesso, o que nos permite replicar essas análises e buscarmos os mesmos resultados (quem sabe eu não escreva um texto sobre isso em breve), além de nos permitir a criação de um fluxo de trabalho completo e automatizado através do Galaxy para facilitar esse processo (sou bioinformata e biólogo computacional, logo, tenho que pensar nisso).
Mas como nem tudo são flores, a previsão e anotação de ORFs potenciais desse artigo foi realizada através do software comercial DNAStar Lasergene, que tem um valor entre $249–$2,399 por ano em sua versão acadêmica e $599–$3,999 em sua versão comercial. Como o artigo cita diretamente a versão DNAStar Lasergene, podemos atribuir o maior valor de cada uma das categoriais.
Mas qual o problema? Simples. Para realmente reproduzir ou replicar esse artigo, meu laboratório ou qualquer laboratório (o que inclui os revisores do artigos) deve desembolsar U$S 2.399,00 por ano por uma licença acadêmica do programa. Multiplicando isso pelo valor do dólar atual (R$ 5,35), teríamos que desembolsar R$ 12.826,90 para reproduzir essa análise. Como o artigo passou por um processo de revisão por pares e suas principais contribuições seriam justamente a montagem do genoma (plenamente reprodutível e replicável), a previsão e anotação das ORFs (reprodutível e replicável somente com uma licença do DNAStar Lasergene), a análise filogenética realizada através dos programas MAFFT, PhyML e MEGA (reprodutíveis e replicáveis pelos softwares serem open source e gratuitos) e o cálculo de identidade de aminoácidos entre as sequências através do MegAlign do DNAStar Lasergene (reprodutível e replicável somente com uma licença do DNAStar Lasergene), devemos aceitar à priori que os revisores eram detentores dessa licença ou que adquiriram no ato da revisão. Se não aceitarmos isso, uma pequena dúvida surgirá em nossa cabeça sobre a validade da metodologia e se realmente ela é reprodutível e válida.
Existiam alternativas open source e gratuitas? Sim, várias. Mas, vou encerrar essa crítica por aqui para não transformar esse texto em uma enorme crítica sobre a utilização de ferramentas com licenças não-permissivas na construção do conhecimento científico. Um texto sobre isso é uma promessa e necessário.
Genômica Estrutural e Funcional
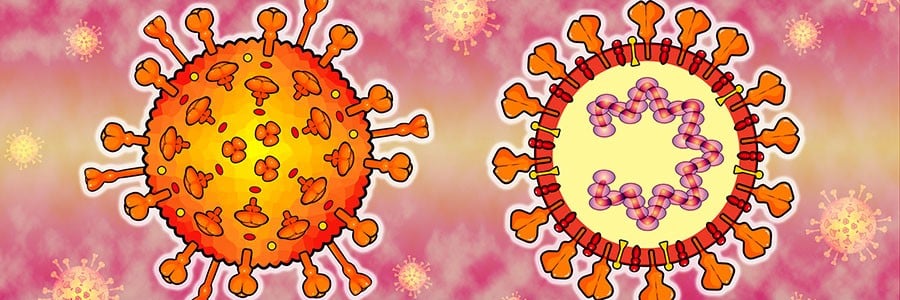
Após sua montagem, observou-se que o genoma do SARS-CoV-2 é composto por uma molécula de RNA de cerca de 30.000 bases de tamanho contendo genes para até 29 proteínas. Ao realizarmos uma comparação rápida, o nosso genoma está na forma de uma dupla hélice de DNA, com o tamanho médio de 3 bilhões de bases e contendo cerca de 19.000–20.000 genes.
Um ótimo infográfico sobre a distribuição estrutura e funcional do SARS-CoV-2 foi apresentado pelo The New York Times.
Em resumo, o SARS-CoV-2 é formado por quatro proteínas estruturais — S , E , M e N — que formam a camada externa do coronavírus e protegem o RNA interno. As proteínas estruturais também ajudam a montar e liberar novas cópias do vírus.
 A proteína spike, ou proteína S, forma picos proeminentes na superfície do vírus, organizando-se em grupos de três. Esses espigões em forma de coroa dão nome aos coronavírus. Parte desse pico pode se estender e se ligar a proteína ACE2, que aparece em células específicas das vias aéreas humanas, permitindo assim que SARS-CoV-2 possa invadir essas células.
A proteína spike, ou proteína S, forma picos proeminentes na superfície do vírus, organizando-se em grupos de três. Esses espigões em forma de coroa dão nome aos coronavírus. Parte desse pico pode se estender e se ligar a proteína ACE2, que aparece em células específicas das vias aéreas humanas, permitindo assim que SARS-CoV-2 possa invadir essas células.
A proteína do envelope, ou proteína E, como o próprio nome diz, é responsável pela formação do envelope viral. Além disso, novas evidências sugerem que essa proteína desempenha outras funções após o vírus invadir uma célula, como se prender às proteínas que ajudam a ativar e desativar nossos próprios genes. Assim, a proteína E pode ser um regulador do padrão de expressão dos nossos genes em uma infecção e atuar como um efetor.
Já a proteína de membrana, ou M, faz parte do revestimento externo do vírus, enquanto a proteína nucleocapsídica, ou N, é responsável pela proteção do RNA viral, mantendo-o estável dentro do vírus. Muitas proteínas N se ligam em uma longa espiral, envolvendo e enrolando o RNA.
Além dessas proteínas estruturais, diversas outras proteínas, como as NPS1–16, desempenham diferentes funções no genoma do SARS-CoV-2, sendo que algumas ainda não apresentam sua função bem definida.
Para um maior detalhamento da genômica estrutural e funcional do SARS-CoV-2, recomendo os artigos de descrição das cepas sequenciadas.
Atualmente, o NCBI apresenta 446 sequências do Genbank e uma sequência do RefSeq do SARS-CoV-2, demonstrando o rápido crescimento dos dados genômicos para esse vírus. Além disso, também estão disponíveis 288 bibliotecas de sequenciamento de nova geração na base de dados do SRA.
Genômica Comparativa
Depois do primeiro sequenciamento do SARS-CoV-2, análises genômicas comparativas começaram a serem realizadas e demostraram que o SARS-CoV-2 pertencia ao grupo dos betacoronavírus e que estava muito próximo do SARS-CoV, responsável por uma epidemia de pneumonia aguda que apareceu em novembro de 2002, na província chinesa de Guangdong e depois se espalhou para 29 países em 2003. Durante esse evento, foram registrados 8.098 casos, incluindo 774 óbitos.
Durante essa epidemia de SARS-CoV, os morcegos do gênero Rhinolophus foram o reservatório desse vírus e um pequeno carnívoro, o civeta da palma (Paguma larvata), pode ter servido como hospedeiro intermediário entre os morcegos e os primeiros casos humanos. Logo, desde essa primeira epidemia de betacoronavírus, evidências sugerem que mesmo surgindo em morcegos, esse vírus teria utilizado um outro organismo como hospedeiro intermediário para a posterior infecção humana. Desde então, muitos betacoronavírus foram descobertos, principalmente em morcegos, mas também em humanos e outros hospedeiros.

Para não estender demais esse texto, vou parar por aqui e discutirei na segunda parte sobre as comparações genéticas do SARS-CoV-2 com outras cepas de diferentes hospedeiros, incluindo os morcegos e o pangolim.
Também falarei sobre a pseudo-hipótese de origem artificial da cepa do COVID-19. Além disso, aproveitarei para discutir sobre a suposta ligação dos casos de COVID-19 com os hábitos alimentares chineses, incluindo a famigerada sopa de morcego.
OBS: Caso encontrem erros, peço que os reporte para correções futuras.
Referências
- Huang, Chaolin & Wang, Yeming & Li, Xingwang & Ren, Lili & Zhao, Jianping & Hu, Yi & Zhang, Li & Fan, Guohui & Xu, Jiuyang & Gu, Xiaoying & Cheng, Zhenshun & Yu, Ting & Xia, Jiaan & Wei, Yuan & Wu, Wenjuan & Xie, Xuelei & Yin, Wen & Li, Hui & Liu, Min & Cao, Bin. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet. 395. 10.1016/S0140–6736(20)30183–5.
- Li, X, Zai, J, Zhao, Q, et al. Evolutionary history, potential intermediate animal host, and cross‐species analyses of SARS‐CoV‐2. J Med Virol. 2020; 1– 10. https://doi.org/10.1002/jmv.25731
- Andersen, K.G., Rambaut, A., Lipkin, W.I. et al. The proximal origin of SARS-CoV-2. Nat Med (2020). https://doi.org/10.1038/s41591-020-0820-9
- Wu, F., Zhao, S., Yu, B. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020). https://doi.org/10.1038/s41586-020-2008-3
- Hatje, Klas & Mühlhausen, Stefanie & Simm, Dominic & Kollmar, Martin. (2019). The Protein‐Coding Human Genome: Annotating High‐Hanging Fruits. BioEssays. 41. 1900066. 10.1002/bies.201900066.
- Li, Wendong & Shi, Zhengli & Yu, Meng & Ren, Wuze & Smith, Craig & H. Epstein, Jonathan & Wang, Hanzhong & Crameri, Gary & Hu, Zhihong & Zhang, Huajun & Zhang, Jianhong & Barr, Jennifer & Field, Hume & Daszak, Peter & Eaton, Bryan & Zhang, Shuyi & Wang, Lin-Fa. (2005). Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science (New York, N.Y.). 310. 676–9. 10.1126/science.1118391.
- Menachery, V., Yount, B., Debbink, K. et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med 21, 1508–1513 (2015). https://doi.org/10.1038/nm.3985
Cite esse artigo
- Menegidio, F. (2019). A origem do SARS-CoV-2 — Parte 1. [Blog] Universo Racionalista. Available at: https://universoracionalista.org/a-origem-do-sars-cov-2-parte-1/
[Accessed 05 Abr. 2020].



