Por Philip Hugenholtz e Soo Jen Low
Publicado no The Conversation
Uma pesquisa publicada dia 25 na Nature Microbiology identificou 54.118 espécies de vírus que vivem no intestino humano – 92 por cento dos quais eram desconhecidos.
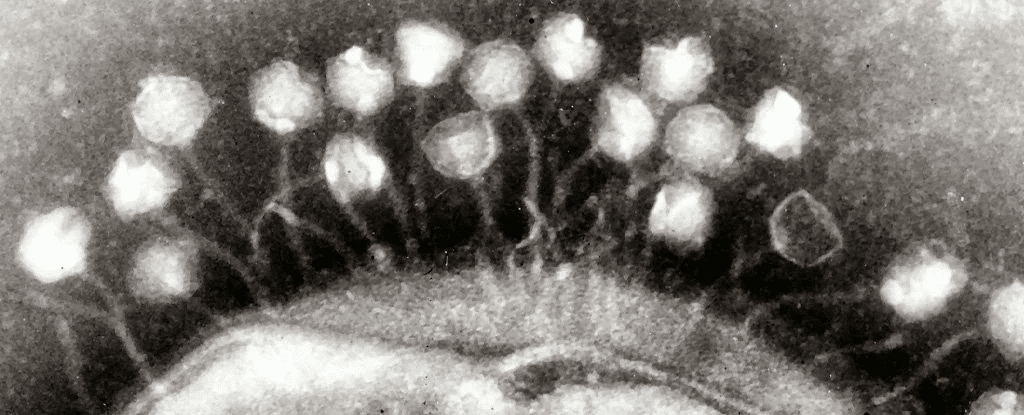
Mas, como nós e nossos colegas do Joint Genome Institute e da Universidade Stanford, na Califórnia, Estados Unidos, descobrimos, a grande maioria deles eram bacteriófagos, ou simplesmente “fagos”. Esses vírus “comem” bactérias e não podem atacar as células humanas.
Quando a maioria de nós pensa em vírus, pensamos em organismos que infectam nossas células com doenças como caxumba, sarampo ou, mais recentemente, COVID-19. No entanto, há um grande número desses parasitas microscópicos em nossos corpos – principalmente em nosso intestino – que têm como alvo os micróbios que vivem lá.
Todo mundo faz cocô (mas nem todo cocô é igual)
Recentemente, tem havido muito interesse no microbioma intestinal humano: a coleção de microrganismos que vivem em nosso intestino.
Além de nos ajudar a digerir nossa comida, esses micróbios têm muitas outras funções importantes. Eles nos protegem contra bactérias patogênicas, modulam nosso bem-estar mental, preparam nosso sistema imunológico quando somos crianças e têm um papel contínuo na regulação imunológica na idade adulta.
É justo dizer que o intestino humano é agora o ecossistema microbiano mais bem estudado do planeta. No entanto, mais de 70 por cento das espécies microbianas que vivem lá ainda precisam ser cultivadas em laboratório.
Sabemos disso porque podemos acessar os esquemas genéticos do microbioma intestinal por meio de uma abordagem conhecida como metagenômica. Esta é uma técnica poderosa pela qual o DNA é extraído diretamente de um ambiente e sequenciado aleatoriamente, dando-nos uma imagem momentânea do que está presente nele e o que ele pode estar fazendo.
Estudos metagenômicos revelaram o quão longe ainda temos que ir para catalogar e isolar todas as espécies microbianas no intestino humano – e ainda mais longe quando se trata de vírus.
11.810 amostras de cocô
Em nossa nova pesquisa, nós e nossos colegas analisamos computacionalmente sequências virais de 11.810 metagenomas fecais disponíveis publicamente, retirados de pessoas em 24 países diferentes. Queríamos ter uma ideia de até que ponto os vírus fixaram residência no intestino humano.
Esse esforço resultou no catálogo Metagenomic Gut Virus, o maior desse tipo até hoje. Este catálogo compreende 189.680 genomas virais que representam mais de 50.000 espécies virais distintas.
Notavelmente (mas talvez previsivelmente), mais de 90 por cento dessas espécies virais são novas para a ciência. Elas codificam coletivamente mais de 450.000 proteínas distintas – um enorme reservatório de potencial funcional que pode ser benéfico ou prejudicial para seus hospedeiros microbianos e, por sua vez, humanos.
Também pesquisamos subespécies de diferentes vírus e descobrimos que alguns mostraram padrões geográficos impressionantes nos 24 países pesquisados.
Por exemplo, uma subespécie do recém-descrito e enigmático crAss-fago era prevalente na Ásia, mas era rara ou ausente em amostras da Europa e América do Norte. Isso pode ser devido à expansão localizada deste vírus em populações humanas específicas.
Uma das funções mais comuns que descobrimos em nossa viagem de campo molecular foram retroelementos geradores de diversidade (DGRs). Trata-se de uma classe de elementos genéticos que sofrem mutação em genes-alvo específicos para gerar variações que podem ser benéficas para o hospedeiro. No caso de DGRs em vírus, isso pode ajudar na corrida armamentista evolutiva em curso contra seus hospedeiros bacterianos.
Curiosamente, descobrimos que um terço das proteínas codificadas por vírus mais comuns têm funções desconhecidas, incluindo mais de 11.000 genes distantemente relacionados a “betalactamases“, que permitem resistência a antibióticos como a penicilina.
Vinculando vírus intestinais a seus hospedeiros microbianos
Tendo identificado os fagos, nossa próxima tarefa foi ligá-los a seus hospedeiros microbianos. CRISPRs, mais conhecidos por suas muitas aplicações na edição de genes, são sistemas imunológicos bacterianos que “lembram” infecções virais passadas e evitam que elas voltem a acontecer.
Eles fazem isso copiando e armazenando fragmentos do vírus invasor em seus próprios genomas, que podem então ser usados para rastrear e destruir especificamente o vírus em encontros futuros.
Usamos esse registro de ataques anteriores para vincular muitas das sequências virais a seus hospedeiros no ecossistema intestinal. Sem surpresa, espécies virais altamente abundantes foram associadas a espécies bacterianas altamente abundantes no intestino, principalmente pertencentes aos filos bacterianos Firmicutes e Bacteroidetes.
Então, o que podemos fazer com todas essas novas informações? Uma aplicação promissora de um inventário de vírus intestinais e seus hospedeiros é a terapia fágica. A terapia fágica é um conceito antigo anterior aos antibióticos, em que os vírus são usados para alvejar seletivamente patógenos bacterianos a fim de tratar infecções.
Tem havido discussão sobre a possibilidade de customizar os microbiomas intestinais das pessoas usando intervenções dietéticas, probióticos, prebióticos ou mesmo transplantes de microbiota fecal, para melhorar a saúde de um indivíduo.
A terapia fágica pode ser um acréscimo útil a esse objetivo, adicionando precisão em nível de espécie ou mesmo subespécie à manipulação do microbioma. Por exemplo, o patógeno bacteriano Clostridioides difficile (ou Cdiff, para abreviar) é uma das principais causas de diarreia adquirida em hospital que poderia ser especificamente alvejada por fagos.
Uma manipulação mais sutil de populações de bactérias não patogênicas no intestino pode ser alcançada por meio de terapia fágica. Um compêndio completo de vírus intestinais é um primeiro passo útil para esses objetivos aplicados.
É importante notar, no entanto, que as projeções de nossos dados sugerem que investigamos apenas uma fração da diversidade viral intestinal total. Portanto, ainda temos um longo caminho a percorrer.



