Vamos seguindo com nossa coleção de textos sobre a A origem do SARS-CoV-2. De início, eu havia previsto apenas 3 partes para essa coleção, mas descobri que existem tantas informações importantes e interessantes sobre a origem desse vírus que resolvi não me limitar.
Na primeira parte, apresentei alguns detalhes sobre os primeiros casos de COVID-19, discuti o sequenciamento da primeira cepa isolada do SARS-CoV-2 e forneci algumas informações sobre sua genômica estrutural e funcional, além das primeiras comparações genômicas com a cepa do SARS-CoV. Na segunda parte, discuti a pseudohipótese de que a atual cepa do SARS-CoV-2 teria ligações com o vírus recombinante produzido em laboratório em 2015 e demonstrei que essa informação, como esperado, não era mais nada do que a boa e velha teoria da conspiração. Na terceira parte, apresentei algumas informações básicas sobre as evidências evolutivas do SARS-CoV-2 e detalhes sobre suas taxas de mutação, e como isso pode afetar na produção de uma vacina.
Agora, nessa quarta parte, vamos voltar a falar sobre as comparações genômicas realizadas em diferentes artigos e quais as evidências filogenéticas que essas nos trouxeram.
Novas comparações genômicas
Como apresentei na primeira parte dessa coleção, após o sequenciamento da primeira cepa isolada do SARS-CoV-2, análises de comparação genômica foram realizadas por diferentes grupos de pesquisa e aplicando diferentes metodologias de bioinformática.
Entre essas primeiras análises realizadas foi observador de forma unânime que o SARS-CoV-2 pertencia ao grupo dos betacoronavírus e que estava muito próximo do SARS-CoV, responsável por uma epidemia de pneumonia aguda que teve início em 2002. Conforme esses dados moleculares, os morcegos do gênero Rhinolophus foram o reservatório primário desse vírus e um pequeno carnívoro, o civeta da palma (Paguma larvata), pode ter servido como hospedeiro intermediário entre os morcegos e os primeiros casos humanos.
 Mas afinal, o que realmente significa um reservatório?
Mas afinal, o que realmente significa um reservatório?
Um reservatório é definido como uma ou mais espécies de animais que não são ou são pouco sensíveis ao vírus, e que hospedarão naturalmente um ou várias cepas desse vírus. A ausência de sintomas da doença é explicada pela efetividade de seu sistema imunológico, o que lhes classifica como ótimos hospedeiros primários e intermediários.
Em fevereiro de 2020, cientistas descobriram em um pangolim da Malásia (Manis javanica) uma cepa viral bastante próxima do SARS-CoV-2, apresentando 99% de concordância genômica, o que poderia sugerir que os pangolins seriam um reservatório mais provável do que os morcegos.

Em março de 2020, outro trabalho voltou a estabelecer uma concordância genômica de 99% entre a cepa do SARS-CoV-2 e a a cepa proveniente de pangolim. Além disso, esse mesmo trabalho observou que o SARS-CoV-2 apresentava uma similaridade de sequências de 96% com a cepa RaTG13, isolada de um vírus de morcego, mas não apresenta mais de 80% de homologia com outros isolados do coronavírus do tipo SARS de outros morcego.
No entanto, um estudo posterior demostrou que o genoma do coronavírus isolado do pangolim seria menos semelhante ao SARS-CoV-2, com apenas 90% de concordância genômica. Essa informação voltou a ser corroborada em outro estudo, que apresentou uma similaridade genômica média de 91% entre a cepa do pangolim e o SARS-CoV-2. Além disso, essa mesma análise corroborou a observação anterior de similaridade de sequências de 96% com a cepa RaTG13 de morcego.
Um acréscimo importante desse estudo foi a disponibilização de uma árvore filogenética abrangente baseado em dados genômicos e no gene RNA-dependent RNA polymerase (RdRp), demonstrando a concordância filogenética dessas estratégias e das previsões evolutivas para o SARS-CoV-2.
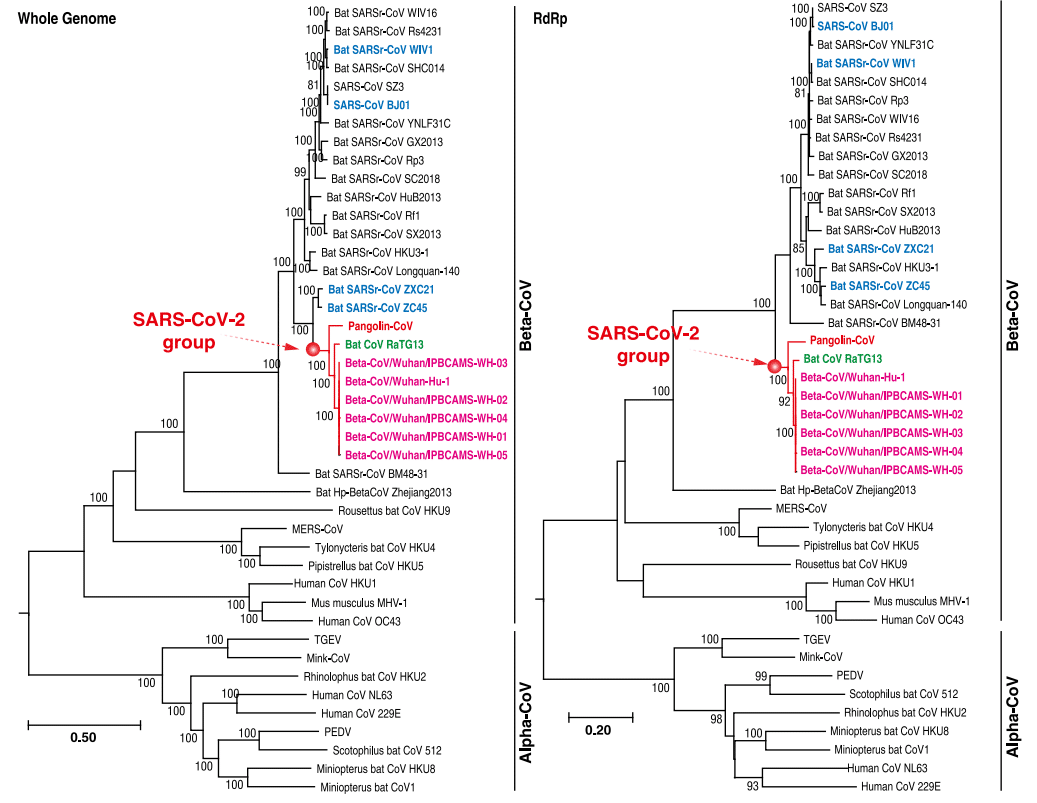
Essas evidências também nos indicariam que o vírus isolado no pangolim também não seria diretamente o responsável pela epidemia de COVID-19 que está ocorrendo atualmente. Além disso, com base nos dados obtidos em literatura, e na árvore filogenética acima, as cepas RaTG13 (morcego) e Pangolin-CoV (pangolim) seriam as cepas disponíveis até o momento com maior similaridade com o SARS-CoV-2, com 96% e 91% respectivamente.
Certo. Aparentemente temos um sério problema para definir a origem do SARS-CoV-2, já que um estudo afirma que essa cepa tem 99% de similaridade com uma cepa de pangolim enquanto outro afirma que essa similaridade é de apenas 90–91%. Além disso, ainda temos a cepa RaTG13, proveniente de um morcego, com 96% de similaridade. Como confiar nesses resultados? Será que temos uma explicação mais precisa sobre a origem desse vírus?
Recombinação genética
As diferenças de similaridade encontradas entre o vírus SARS de pangolim com o SARS-CoV-2 possui uma explicação. O coronavírus isolado do pangolim apresenta uma semelhança de 99% com o SARS-CoV-2 em uma região específica da proteína spike, ou S, que corresponde aos 74 aminoácidos envolvidos no domínio de ligação ao receptor da ACE2 (enzima de conversão da angiotensina 2), aquele que permite a entrada do vírus células em humanas para infectá-los. Por outro lado, o vírus RaTG13 isolado do morcego R. affinis é altamente divergente nessa região específica, apresentando apenas 77% de similaridade. Isto significa que o coronavírus isolado do pangolim é capaz de entrar nas células humanas, enquanto o isolado do morcego R. affinis não é.
Esses detalhes são bem apresentados nos comentários da Carta ao Editor abaixo:
Uma pesquisa BLAST das sequências genômicas de SARS-CoV-2 sugeriu que os coronavírus intimamente relacionados eram bat/RaTG13 e Pangolin/1, com ∼96% e ∼90,5% de identidade geral das sequências genômicas, respectivamente.
Nos genes 1ab, S, E, M e N, o coronavírus bat/RaTG13 exibiu 96,2%, 97,3%, 100%, 99,6% e 99,0% de aminoácidos idênticos ao de SAR-CoV-2, respectivamente, enquanto o coronavírus Pangolin/1 mostrou 96,3%, 92,4%, 100%, 98,7% e 97,9% de os aminoácidos idênticos aos de SARS-CoV-2, respectivamente.
No entanto, foi notável que a identidade da sequência do gene 1b do coronavírus pangolin/1 foi maior que o coronavírus RaTG13 de origem em morcego, sendo o mais alto 99,3%.
Assim, essas comparações genômicas sugerem que o vírus SARS-Cov-2 é o resultado de uma recombinação entre dois vírus diferentes, um próximo ao RaTG13 e outro mais próximo ao Pangolin-CoV. Em outras palavras, é uma quimera entre dois vírus pré-existentes.
Esse mecanismo de recombinação já havia sido descrito nos coronavírus, em particular para explicar a origem do SARS-CoV. É importante saber que a recombinação resulta em um novo vírus potencialmente capaz de infectar uma nova espécie hospedeira. Para que a recombinação ocorra, os dois vírus divergentes devem ter infectado o mesmo organismo simultaneamente.
Duas perguntas permanecem sem resposta: em qual organismo essa recombinação ocorreu? (um morcego, um pangolim ou outra espécie?) E, acima de tudo, sob que condições essa recombinação ocorreu? Essas são resposta que ainda não temos e só existirão em um futuro texto quando a comunidade científica as descobrir.
Então, vou parar por aqui novamente. Ainda não sei qual será o tema do próximo texto, mas ainda temos alguns assuntos para discutir sobre a Origem do SARS-CoV-2.
Entre esses assuntos, temos a segunda pseudo-hipótese de que o SARS-CoV-2 possa ter escapado de um laboratório na China. Será que isso é realmente possível? E a sopa de morcego? É uma hipótese para essa pandemia?
OBS: Caso encontrem erros, peço que os reporte para correções futuras.
Referências
- Li, Wendong & Shi, Zhengli & Yu, Meng & Ren, Wuze & Smith, Craig & H. Epstein, Jonathan & Wang, Hanzhong & Crameri, Gary & Hu, Zhihong & Zhang, Huajun & Zhang, Jianhong & Barr, Jennifer & Field, Hume & Daszak, Peter & Eaton, Bryan & Zhang, Shuyi & Wang, Lin-Fa. (2005). Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science (New York, N.Y.). 310. 676–9. 10.1126/science.1118391.
- Menachery, V., Yount, B., Debbink, K. et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med 21, 1508–1513 (2015). https://doi.org/10.1038/nm.3985
- Liu, P.; Chen, W.; Chen, J.-P. Viral Metagenomics Revealed Sendai Virus and Coronavirus Infection of Malayan Pangolins (Manis javanica). Viruses 2019, 11, 979.
- Wang, C, Liu, Z, Chen, Z, et al. The establishment of reference sequence for SARS‐CoV‐2 and variation analysis. J Med Virol. 2020; 1– 8. https://doi.org/10.1002/jmv.25762
- Xiao, Kangpeng & Zhai, Junqiong & Feng, Yaoyu & Zhou, Niu & Zhang, Xu & Zou, Jie-Jian & Li, Na & Guo, Yaqiong & Li, Xiaobing & Shen, Xuejuan & Zhang, Zhipeng & Shu, Fanfan & Huang, Wanyi & Li, Yu & Zhang, Ziding & Chen, Rui-Ai & Wu, Ya-Jiang & Peng, Shi-Ming & Huang, Mian & Shen, Yongyi. (2020). Isolation and Characterization of 2019-nCoV-like Coronavirus from Malayan Pangolins. 10.1101/2020.02.17.951335.
- Zhang, Tao & Qunfu, Wu & Zhang, Zhigang. (2020). Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Current Biology. 10.1016/j.cub.2020.03.022.
- Zhang, Jiahao & Jia, Weixin & Zhu, Junhai & Li, Bo & Xing, Jinchao & Liao, Ming & Qi, Wenbao. (2020). Insights into the cross-species evolution of 2019 novel coronavirus. Journal of Infection. 10.1016/j.jinf.2020.02.025.
- Graham, Rachel & Baric, Ralph. (2009). Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. Journal of virology. 84. 3134–46. 10.1128/JVI.01394–09.
Cite esse artigo
Menegidio, F. (2019). A origem do SARS-CoV-2 — Parte 4. [Blog] Universo Racionalista. Available at: https://universoracionalista.org/a-origem-do-sars-cov-2-parte-4/
[Accessed 05 Abr. 2020].



