Vamos prosseguir com nossa coleção de textos sobre a A origem do SARS-CoV-2. Na primeira parte, apresentei alguns detalhes sobre os primeiros casos de COVID-19, discuti o sequenciamento da primeira cepa isolada do SARS-CoV-2 e forneci algumas informações sobre sua genômica estrutural e funcional, além das primeiras comparações genômicas com a cepa do SARS-CoV. Na segunda parte, discuti a pseudohipótese de que a atual cepa do SARS-CoV-2 teria ligações com o vírus recombinante produzido em laboratório em 2015 e demonstrei que essa informação, como esperado, não era mais nada do que a boa e velha teoria da conspiração.
Nessa terceira parte, antes de prosseguir com as comparações genômicas, vamos discutir rapidamente o que sabemos sobre a evolução do SARS-CoV-2 e o motivo dessas informações serem importantes.
Evolução genética do COVID-19
Quando ouvimos o termo árvore evolutiva, com certeza pensamos na única imagem presente no livro Origem das espécies, de Charles Darwin, e no estudo das relações entre diferentes espécies ao longo de milhões de anos. Embora o conceito de árvore evolutiva tenha realmente surgido nesse livro, ele vem sendo utilizado na biologia desde então, podendo ser aplicado a qualquer coisa que evolua, incluindo os vírus.

Quando pensamos em vírus, uma pergunta extremamente complexa nos surge na cabeça: eles são seres vivos? E nesse momento, uma das questões mais antigas da biologia retorna para o centro de diversos debates acirrados.
Durante muito tempo, um consenso científico estabeleceu que os vírus não seriam seres vivos por não crescerem e não conseguirem se reproduzir fora de uma célula, necessitando de toda a maquinaria molecular do hospedeiro para criar cópias de si mesmo. No entanto, nos últimos 15 anos, o surgimento de vírus gigantes encontrados em amebas, denominados mimivírus e megavírus, complicou essa visão dominante de que os vírus são simples estruturas não-vivas. A descoberta de que alguns desse vírus gigantes poderiam conter mais genes do que algumas bactérias simples, e de que esses genes poderiam ser encontradas nos domínios Archaea, Bacteria e Eucarya, tornou o debate sobre a classificação dos vírus novamente relevante e cinzento na comunidade científica. Sendo que atualmente, um número crescente de pesquisadores vem defendendo a criação de um quarto domínio para comportar os vírus.
No entanto, indiferente da classificação dos vírus como seres vivos ou estruturas não-vivas, seu estudo evolutivo permite que os pesquisadores aprendam mais sobre como seus genes funcionam e possibilita a realização de inferências sobre a propagação do vírus em todo o mundo e que tipo de vacina pode ser mais eficaz para combatê-lo.
Como as sequências evoluem?
Todos os vírus, o que incluí o SARS-CoV-2, evoluem por mutações. Ou seja, observamos mudanças em seu código genético ao longo do tempo. Esse é um fato muito bem estabelecido, indiferente se as pessoas aceitam ou não. Os vírus e o surgimento de novas cepas virais são extremamente bem explicados com base na Teoria Sintética da Evolução.
Não pretendo nesse texto discorrer sobre todos os processos evolutivos envolvidos no surgimento de novas cepas virais, mas essas informações estarão dissolvidas ao longo dos textos, quando necessárias.
O processo como as mutações ocorrem nos vírus, assim como em outros organismos (vou utilizar a palavra de uma forma ampla), se assemelha bastante com uma brincadeira de telefone sem fio.
O primeiro participante pode começar a brincadeira com a palavra inglesa BEAST. Ele pode sussurrar para a segunda pessoa que entende a palavra de forma completa e passa para a próxima. A terceira pessoa pode acidentalmente ouvir a palavra como FEAST, provocando uma substituição de letra. Passando o termo para o próximo, o quarto individuo pode entender a palavra BREAST, gerando uma inserção na palavra original. A quinta pessoa pode entender BEST, deletando uma letra na palavra original. Já a sexta pessoa pode entender BEATS, gerando uma inversão nas duas últimas letras da palavra original. Esse processo pode continuar até o último participante e teremos um resultado interessante, com a conservação da palavra original por alguns participantes, o surgimento de novas palavras funcionais, o surgimento de amontoados de letras que não apresentam um significado em conjunto ou mesmo o ressurgimento da palavra original a partir de uma variante.
Esse processo é bem descrito na imagem abaixo:
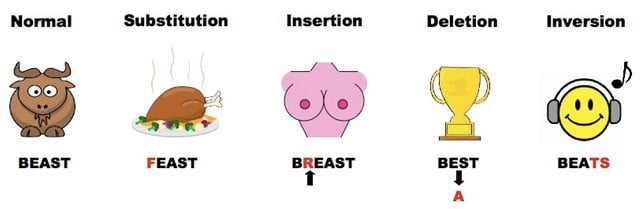
Muito parecido com o exemplo acima, a sequência do genoma do SARS-CoV-2 muda com o tempo: as mutações ocorrem aleatoriamente e quaisquer alterações que ocorram em um determinado vírus serão herdadas pelas cópias da próxima geração. Buscando estudar como essas mutações ocorrem ao longo do tempo, diversos cientistas vem desenvolvendo uma grande quantidade de modelos de evolução genética para tentar determinar a história evolutiva mais provável do vírus, sendo esse um campo em constante expansão na minha área de atuação, a Bioinformática e a Biologia Computacional.
O sequenciamento de DNA é o processo que permite encontrar experimentalmente a sequência de nucleotídeos (A, C, G e T) — os blocos químicos dos genes — de um determinado pedaço de DNA. O sequenciamento de DNA já é amplamente utilizado na medicina para o estudo de diferentes doenças genética humanas, mas nos últimos anos, ele se tornou parte rotineira do cuidado viral e, à medida que o sequenciamento se torna cada vez mais barato, o sequenciamento viral se torna ainda mais frequente com o passar do tempo.
O RNA é uma molécula semelhante ao DNA, e é essencialmente uma cópia temporária de um pequeno segmento de DNA. Especificamente, no dogma central da biologia , o DNA é transcrito para o RNA. O SARS-CoV-2 é um vírus de RNA, o que significa que nossas tecnologias de sequenciamento de DNA não podem decodificar diretamente sua sequência. No entanto, os cientistas podem primeiro reverter a transcrição do RNA do vírus em DNA complementar (ou cDNA), que pode ser sequenciado.
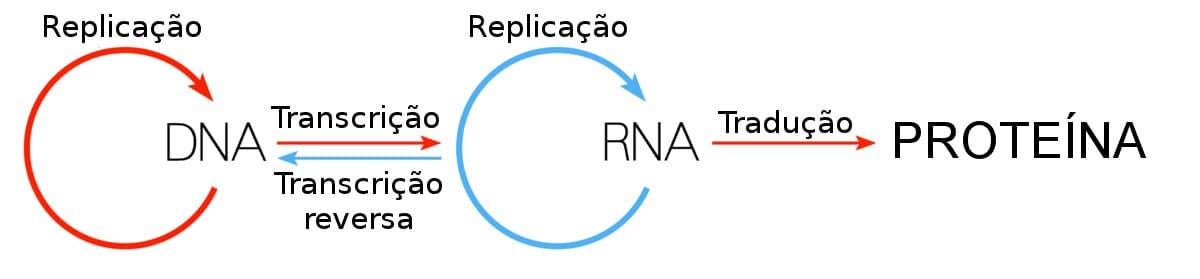
Dada uma coleção de sequências genômicas virais, podemos utilizar diferentes modelos de evolução de sequências para prever a história do vírus e podemos usá-lo para responder perguntas como: “Com que rapidez ocorrem as mutações?” ou “Onde no genoma ocorrem mutações?” Saber quais genes estão sofrendo mutações com frequência pode ser útil no design de medicamentos.
O rastreamento de como os vírus foram alterados em um local também pode responder a perguntas como “Quantos surtos separados existem na minha comunidade?” Esse tipo de informação pode ajudar as autoridades de saúde pública a conter a propagação do vírus.
Para o COVID-19, por exemplo, foi criada uma iniciativa global para compartilhar genomas virais de forma aberta e livre, denominada Gisaid. Dada uma coleção de sequências com datas das amostras, os cientistas podem inferir a história evolutiva das amostras em tempo real e usar as informações para inferir a história das transmissões.
Entre os projetos que surgiram com base nas perspectivas de análise acima, destaca-se o Nextstrain, um projeto de código aberto que fornece aos usuários relatórios em tempo real da propagação da influenza sazonal , Ebola e muitas outras doenças infecciosas. Mais recentemente, o Nextstrain tem liderado o rastreamento evolutivo do COVID-19, fornecendo uma análise em tempo real e um relatório de situação que deve ser legível pelo público em geral, o que inclui a grande mídia de massa. Além disso, o projeto permite que a população global se beneficie de seus esforços traduzindo o relatório da situação para muitos outros idiomas.
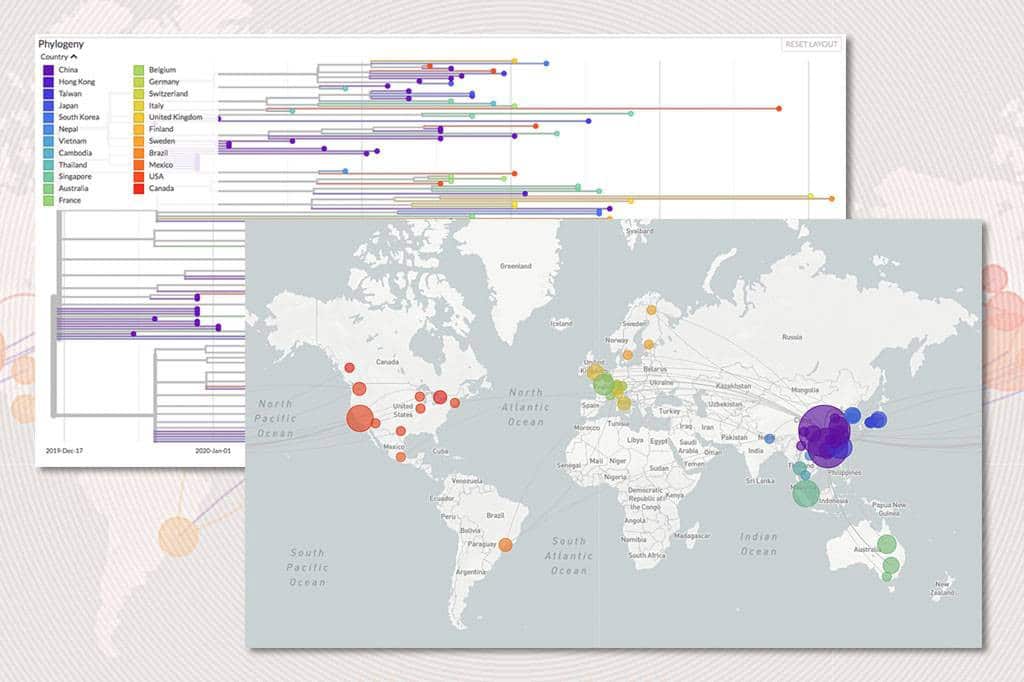
Eu dediquei um texto exclusivo para tratar desses dois projetos e de outros desenvolvidos pela comunidade de Software Livre. Vale a leitura!

O que aprendemos sobre o COVID-19?
Com base nos dados atuais, parece que a taxa de mutações do SARS-CoV-2 é muito mais lenta do que a gripe sazonal. Especificamente, o SARS-CoV-2 parece ter uma taxa de mutação inferior a 25 mutações por ano, enquanto a gripe sazonal tem uma taxa de mutação de quase 50 mutações por ano.
Dado que o genoma do SARS-CoV-2 é quase duas vezes maior que o genoma da gripe sazonal, parece que a gripe sazonal sofre mutação cerca de quatro vezes mais rápida que o SARS-CoV-2. O fato de a gripe sazonal sofrer uma mutação tão rápida é precisamente o motivo pelo qual é capaz de evitar nossas vacinas.
Portanto, a taxa de mutação significativamente mais lenta do SARS-CoV-2 nos dá esperanças para o desenvolvimento potencial de vacinas eficazes e duradouras contra o vírus.
Por enquanto é isso. Prosseguimos nos próximos textos, pois ainda existem muitas informações interessantes para serem ditas sobre o SARS-CoV-2.
OBS: Caso encontrem erros, peço que os reporte para correções futuras.
Referências
- Arslan D, Legendre M, Seltzer V, Abergel C, Claverie JM. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc Natl Acad Sci U S A. 2011;108(42):17486–17491.
- Legendre M, Arslan D, Abergel C, Claverie JM. Genomics of Megavirus and the elusive fourth domain of Life. Commun Integr Biol. 2012;5(1):102–106.
- James Hadfield, Colin Megill, Sidney M Bell, John Huddleston, Barney Potter, Charlton Callender, Pavel Sagulenko, Trevor Bedford, Richard A Neher, Nextstrain: real-time tracking of pathogen evolution, Bioinformatics, Volume 34, Issue 23, 01 December 2018, Pages 4121–4123.
- Klinkenberg D, Backer JA, Didelot X, Colijn C, Wallinga J (2017) Simultaneous inference of phylogenetic and transmission trees in infectious disease outbreaks. PLOS Computational Biology 13(5): e1005495.
Cite esse artigo
- Menegidio, F. (2019). A origem do SARS-CoV-2 — Parte 3. [Blog] Universo Racionalista. Available at: https://universoracionalista.org/a-origem-do-sars-cov-2-parte-3/
[Accessed 07 Abr. 2020].



